Wildtyp-Transthyretin-(ATTRwt-)Amyloidose
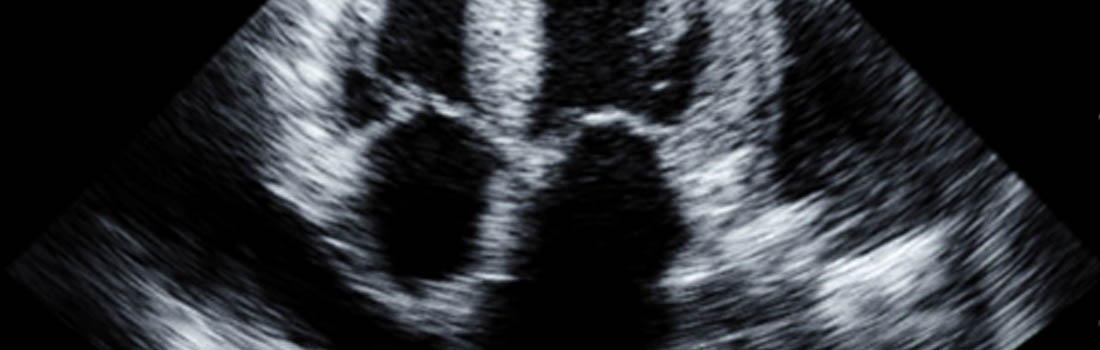
Die ATTRwt-Amyloidose ist durch die Ablagerung von Amyloidfibrillen im Gewebe charakterisiert, die aus dem Eiweiß Transthyretin (Präalbumin) entstanden sind.
Transthyretin wird fast ausschließlich in der Leber gebildet und dient dem Transport des Schilddrüsenhormons sowie von Vitamin A. Zu Amyloidablagerungen kommt es insbesondere bei älteren Männern. Das Herz ist bei dieser Amyloidoseform bei fast allen Patienten betroffen.
Ursachen
Die Ursache der ATTRwt-Amyloidose ist nicht im Detail geklärt. Im Gegensatz zur hereditären Form kommt zur Umwandlung von Transthyretin in Amyloidfibrillen auch ohne Vorliegen einer Mutation im Transthyretingen. Auch wenn bei ca. 20% der Patienten eine monoklonale Gammopathie vorliegt, ist diese nicht ursächlich für die ATTRwt-Amyloidose. Eine Chemotherapie ist bei diesen Patienten mit ATTRwt-Amyloidose nicht wirksam.
Diagnose
Die Frühdiagnose einer ATTRwt-Amyloidose stellt eine besondere Herausforderung dar, da die Veränderungen anfangs auch durch häufigere Erkrankungen, wie zum Beispiel Bluthochdruck, erklärt sein könnten.
Die Diagnose der ATTRwt-Amyloidoseerfordert daher eine sorgfältige klinische und apparative Untersuchung. Die Echokardiographie und die Magnetresonanztomographie des Herzens sind wichtige Untersuchungen, die einen ersten Hinweis auf eine Amyloidose des Herzens ergeben können. Ergänzend ist die Durchführung von Bluttests mit Bestimmung der Herzbiomarker sowie der Nierenfunktion zur Stadieneinteilung sinnvoll. Die definitive Bestätigung erfolgt mittels einer Biopsie mit Färbung durch gezielt gegen Transthyretinamyloid gerichtete Antikörper. Alternativ kann auch eine Knochenszintigraphie die Diagnose ATTR-Amyloidose stellen, wenn eine monoklonale Gammopathie ausgeschlossen ist. Eine genetische Untersuchung ist erforderlich, um die ATTRwt-Amyloidose von der vererblichen Form (ATTRv-Amyloidose) abzugrenzen.
Symptome
Die Symptome von Patienten mit ATTRwt-Amyloidose sind vor allem durch die Zeichen einer Herzinsuffizienz geprägt und stetig progredient.
Da die klinischen Symptome mit Leistungsminderung, Flüssigkeitsansammlung und/oder Herzrhythmusstörungen allerdings unspezifisch sind und bei jeder Form der Herzschwäche auftreten können, wird eine ATTRwt-Amyloidose meistens erst im fortgeschrittenen Stadium erkannt. Dann liegen jedoch bereits erhebliche Organschäden vor. Eine besondere Häufung der ATTRwt-Amyloidose wird bei Patienten mit Carpaltunnelsyndrom, aber auch bei Vorliegen einer Spinalkanalstenose beobachtet.
Therapie
Vyndaqel® (Tafamidis) 61 mg hat von der Europäischen Kommission am 18.02.2020 die Zulassung zur Behandlung der Transthyretin-Amyloidose mit Kardiomyopathie (ATTR-CM) erhalten.
Vyndaqel® (Tafamidis) 61 mg sollte unter der Kontrolle eines in der Behandlung von Patienten mit Amyloidose oder Kardiomyopathie erfahrenen Arztes begonnen werden. Hierbei muss die Diagnose ATTR-CM durch ein mit der Behandlung von Amyloidose oder Kardiomyopathie erfahrener Arzt bestätigt werden und eine AL Amyloidose ausgeschlossen werden, bevor mit der Behandlung mit Vyndaqel® (Tafamidis) 61mg begonnen wird. Hierbei eignen sich folgende Untersuchungsverfahren: Knochenszintigraphie und Blut-/Urin-Untersuchungen und/oder histologische Untersuchung einer Biopsie und Genotypisierung des Transthyretin, um es als Wildtyp oder hereditär zu charakterisieren.
Studien
Ferner sind klinische Studien in mehreren Zentren offen, bei denen die Wirksamkeit von Medikamenten, welche durch Interaktion mit der TTR-mRNA in der Leber die Neubildung von TTR reduziert, untersucht werden.